Quels changements avec le nouveau règlement sur les dispositifs médicaux ?

Initialement prévu pour le 26 Mai 2020, le règlement relatif aux dispositifs médicaux (DM) entrera en application à partir du 26 Mai 2021. Décidé en 2017, ce nouveau règlement couvrira plus de produits et instaure de nouvelles mesures pour les entreprises concernées. Quels sont les changements ? Qui est concerné ? Découvrez les réponses au sein de cet article.
Entré en vigueur le 25 Mai 2017, le nouveau Règlement sur les Dispositifs Médicaux (UE) 2017/745 (également appelé RDM ou EU-MDR) devait être appliqué dès le 26 Mai 2020. Cependant, en raison de la crise sanitaire liée à la COVID-19, la mise en application du règlement a été repoussée afin de permettre aux entreprises concernées de se concentrer sur la gestion de la pandémie.
En quoi consiste ce nouveau règlement ?
Ce règlement a plusieurs objectifs, cependant, l’objectif principal est d’optimiser la sécurité des patients en appliquant des mesures plus strictes, notamment grâce à une évaluation de la conformité des dispositifs médicaux, ainsi qu’une surveillance plus poussée. Le but du durcissement de ces mesures est bien évidemment de s’assurer que tous les produits mis sur le marché ne soient pas dangereux. Ce règlement s’applique à l’Union européenne et remplace la directive sur les dispositifs médicaux (93/42/CEE) en vigueur jusque-là. La précision et l’exigence du nouveau règlement se constatent d’ailleurs par la taille du RDM : 175 pages contre 63 pour l’ancienne directive.
Le RDM a également pour but de permettre une mise à jour totale des règles relatives aux dispositifs médicaux (et de leurs accessoires) tant pour leur mise en marché, que pour leur mise à disposition et leur mise en service. Cette mise à jour des règles porte également sur la conduite des investigations cliniques de ces dispositifs médicaux.
Enfin, le Règlement Européen relatif aux Dispositifs Médicaux permet de cadrer l’industrie des dispositifs médicaux en offrant plus de transparence et de normes. En effet, certaines directives européennes concernant les dispositifs médicaux dataient de 1990 et n’étaient que des directives, ce qui veut dire qu’elles n’étaient pas d’application directe et que chaque état membre de l’UE les a transposées dans sa législation nationale. Cette subtilité offrait donc à chaque état la possibilité de l’interpréter comme il le souhaite. A l’inverse, le RDM est d’application directe et concerne tous les états membres. Il va donc forcément créer une uniformisation des obligations relatives aux dispositifs médicaux.
Quels sont les changements apportés par le RDM ?
Le RDM va donc apporter plusieurs mesures permettant de renforcer la sécurité des patients. Les principaux changements sont les suivants :
- Plus de produits couverts par le RDM : Plus de produits seront couverts par ce nouveau règlement que par la directive précédente. En effet, les dispositifs « sans finalité médicale » (également appelés dispositifs esthétiques) sont désormais concernés par le règlement. On retrouve entre autres, parmi ces produits les lentilles de contact « fantaisie », les produits destinés à être introduits dans le corps en vue de modifier l’anatomie, les substances injectées pour un comblement pour la peau, les muqueuses ou le visage, les prothèses mammaires, les équipements utilisés pour réduire, enlever ou détruire des tissus adipeux, etc.).
- Plus de règles de classification : Dans la directive sur les dispositifs médicaux (93/42/CEE), le nombre de règles de classification est de 18, le RDM en impose 22. La classe des différents dispositifs médicaux dépend des risques qu’ils présentent et de leur destination. Les différentes Classes sont les suivantes : I, IIa, IIb, III. Les produits avec les risques les plus élevés sont ceux de la Classe III, et ceux présentant un risque moins élevé sont ceux de la Classe I. La Classe I dispose d’ailleurs de 3 sous-classes : la Classe Is (Produits vendus à l’état stérile), la Classe Im (les produits présentant une fonction de mesure) et la Classe Ir (les produits pouvant être réutilisés avec un nettoyage).
- Amélioration de l’évaluation avant la mise en marché : Afin d’optimiser la sécurité des patients, de nouvelles règles d’évaluation des dispositifs médicaux avant leur mise en marché ont été instaurées. Ainsi, plus de données et d’informations sur les produits devront être fournies par le fabricant pour la mise en marché de ses dispositifs médicaux. Par ailleurs, afin d’harmoniser le niveau d’exigence, les organismes notifiés des différents états membres de l’UE sont placés sous contrôle Européen. Ces organismes notifiés distribueront les certifications de marquage CE pour les dispositifs médicaux. Pour ce faire, des inspections (parfois non-programmées) auront lieu dans les locaux des fabricants de DM.
- Amélioration de l’évaluation des données cliniques : La collecte des données cliniques relatives aux dispositifs médicaux a été renforcée. Ainsi, les règles liées à la collecte de ces données s’alignent sur celles relatives aux essais cliniques concernant les médicaments (protection des sujets vulnérables, consentement éclairé, etc.). Les études cliniques effectuées dans au moins 2 pays européens devront faire l’objet d’une évaluation coordonnée unique.
- Mise en place d’EUDAMED : EUDAMED est une base de données européenne sur les dispositifs médicaux. Son objectif est d’informer les patients, les professionnels de santé et le grand public des informations relatives aux DM. Cette base de données permettra de concentrer au même endroit, les informations concernant la surveillance du marché, les investigations cliniques, la vigilance, etc.
- Amélioration du suivi post-commercialisation : Les fabricants devront mettre en place un nouveau système de gestion de la qualité ainsi qu’un système de surveillance des dispositifs médicaux après commercialisation. Cette surveillance s’effectuera par l’intermédiaire de rapports réguliers d’activité de sécurité des dispositifs médicaux. Un outil de suivi à l’échelle européenne sera mis en place prochainement.
Les mesures citées précédemment ne représentent qu’une partie des changements dus au règlement. Le Règlement relatif aux Dispositifs Médicaux regroupe toutes les mesures à prendre de manière détaillée.
Les principales dates du RDM
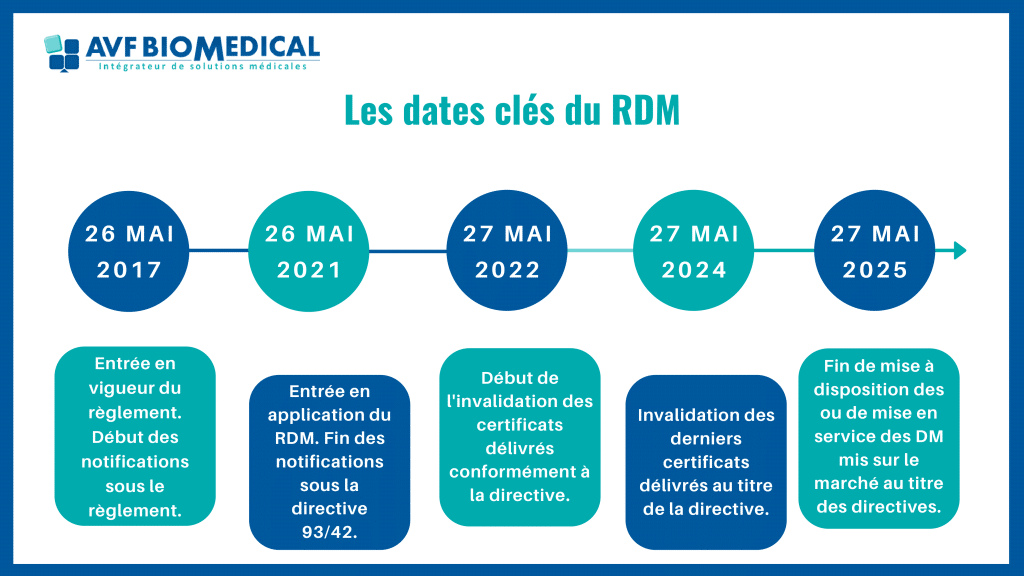
L’entrée en application de ce règlement est fixée au 26 mai 2021, cependant sa mise en œuvre s’étale sur plusieurs années :
- 26 Mai 2021 : Entrée en application, les dispositifs médicaux de Classe I doivent être en conformité avec le RDM.
- 2022 – 2024 : Invalidation des certificats conformes aux directives précédent le RDM.
- 27 Mai 2024 : Tous les dispositifs médicaux de Classe IIa à III doivent être conformes.
- 27 Mai 2025 : Fin de mise à disposition ou de mise en service ses DM mis sur le marché au titre des directives.